Skeletal Dysplasias
In her classic children’s book, The Tale of the Flopsy Bunnies, Beatrix Potter began the story with the following sentence: “It is said that the effect of eating too much lettuce is soporific”. She might well have been talking about the attitude most physicians have toward the study of skeletal dysplasias. For many medical students, the usual LD-50 for a chapter on dysplasia is about 1 – 2 minutes.
Although many dysplasias are quite rare, some occur with sufficient frequency that most general practice radiologists will see them and should know something about them. This chapter gives an extremely simplified meatball approach to the diagnosis of these more common disorders. You won’t find rare syndromes such as bird-headed dwarvism on the differentials listed herein.
I first became aware of the importance of skeletal dysplasias during my first pediatric rotation in medical school. This rotation was in a pediatrics special care unit through which all the dysplasia work-ups in our medical center were funnelled. In my month on that service, I saw more unusual physical findings than I had ever imagined possible. Many of the disorders I saw that month were so rare that I had not only never heard of them, but have never seen them since then. My initial reaction was that I was wasting a month that could be better spent seeing garden variety pediatric complaints.
However, one particular child made the whole month-long rotation worthwhile to me. This little one was a month old and was extremely challenging for a second year medical student to examine. There was at least one unusual finding in every single part of this baby’s physical exam. This child had been born in Mexico, where they have some pretty smart doctors. These physicians certainly realized that this child had an unusual syndrome outside of their experience, did their best to figure it out, and told the parents that the child would be massively mentally retarded. The parents were heartbroken, and decided to seek a second opinion in a Major Medical Center in the U. S. They were given some other diagnosis at that MMC, which was also associated with mental retardation. The parents decided to give it one more try, and came to our MMC for a third opinion. I examined the infant and presented her to the attending. He listened to my differential diagnosis, and then re-examined the child. He then carefully explained to us why my list was wrong and why all of the diagnoses ventured by experts elsewhere were wrong. He summed it up by stating, “This child can only have the Hallerman-Streiff syndrome”. This didn’t mean much to me until our attending pointed out that Hallerman-Streiff syndrome was associated with many things, but not with extensive mental retardation or a shortened life. This much more hopeful prognosis gave the parents great joy, and my respect for the power of a third opinion went way up.
This experience did not make me want to go sign up for a dysplasia fellowship. But, it did teach me that sometimes giving a good prognosis is one of the best things you can do for a patient. A diagnosis, therefore, may be of great utility, even though it leads to no change in the patient’s treatment. It also allows the patient to receive appropriate genetic counseling, which can be very valuable indeed.
In the years since then, I have seen the radiographs of quite a few dysplasia patients. This experience has given me a pretty good idea of which syndromes most other radiologists are going to encounter in their practices. This has led to the following practical approach:
A Quick & Dirty Approach to Skeletal Dysplasias
My personal approach has these five simple steps:
- The first thing is to somehow tumble to the idea that the patient might have a dysplasia.
- Next, check and see if the findings are due to some acquired problem that you already know a lot about — if so, life is good.
- OK, sigh… it’s not an acquired disorder. Run through your mental short list of commonly seen congenital dysplasias — if this patient is on it, life can still be good.
- If the patient isn’t on the short list, look for the child in Taybi and Lachman’s seminal book.
- If you are still uncertain, send the patient to an expert for another opinion. Even if you are pretty sure of the diagnosis, this still may not be a bad rule.
1. Could it be a dysplasia?
This first step may be the hardest one. There are a lot of physicians out there in deep denial about dysplasias. They dislike so much the idea of having to do a dysplasia workup that they may refuse to even put dysplasia on their mental lists. As soon as they consider the “D” word, up come visions of bird-headed dwarves and hand-foot-uterus syndrome. I call this the mental Moro reflex. My advice: get over it — bird-headed dwarvism and hand-foot-uterus syndrome are quite rare (I’m still waiting to see my first case of either). Most cases of dysplasia that I see are usually due to some common acqured disorder or some relatively common congenital dysplasia.
To be a bit more charitable to my colleagues, sometimes we also tend to focus so tightly on one particular finding and how to explain it that we miss the big picture. One handy clue to recognizing dysplasias is to note that the process is systemic or generalized, rather than focal. Another handy clue is to recall what the word dysplasia really means. Dysplasia comes from Latin roots dys and plasia, meaning “bad growth”. Therefore, if your patient has bones that are oddly-shaped in some way, they are probably dysplastic. As you look at the shape of a bone, if you find yourself thinking words like “strange”, “bizarre”, “peculiar”, “odd”, etc. train your brain to automatically translate this into dysplasia, dysplasia, dysplasia,….
2. Could this be an acquired process?
Don’t forget that there are also acquired causes of dysplastic bones, of course. This simple fact saves my personal butt about half the time when I’m asked to consult on someone with unusual-looking bones. Acquired disorders are much more common than congenital dysplasia in the patient population I usually see, even in the big tertiary medical center in which I practice. Not only that, but unusual manifestations of common, acquired disorders are still more common than congenital dysplasias in my practice. Unless you end up practicing in some large center that specialized in dysplasias, your experience will probably be a lot like mine.
In order for growing bones to end up with a normal shape and size, they need normal muscle pull and gravitational loading while they are growing. Therefore, any process that interferes with this normal loading may lead to odd-looking bones. Paraplegia or quadriplegia are extreme examples of this. Childhood paraplegics may end up with extremely gracile (long and thin) bones, with coxa valga, a small and odd-looking pelvis, and very little muscle mass. On the other end of the spectrum, any chronic disease that keeps a child bedridden a lot during their growth phase (leukemia, juvenile chronic arthritis, hemophilia, etc.) may give them mildly dysplastic bones that look like one of the neuromuscular diseases, such as paraplegia. Fractures that heal poorly or which have had extensive surgery can appear quite bizarre at times, and should be on the differential for acquired bone dysplasias. So, this is where history comes in handy. Once you have gotten the clinical lowdown, you should be able to refine the diagnosis a bit more.
3. Could this be a common dysplasia?
Specialty books list hundreds of different types and subtypes of skeletal dysplasias. This is surely heady wine for the connoisseur of such things. However, the rest of us would love to have a short list of the ones we’re most likely to see. Some guidance can be gained from a recent compilation by Kozlowski and Beighton. These authors listed every dysplasia that they could think of and tried to rank each from 0 to 4 stars, corresponding to how common it was. A four star ranking meant that there are over 1000 cases of it reported in the literature. Three stars means that there are between 100 and 1000 cases, and so on. I agree with most of their rankings, but would adjust some of the entities a bit. My modified Short List Of Dysplasias (SLOD) is listed here in alphabetical order:
- Achondroplasia
- Cleidocranial dysostosis
- Dactyly
- Brachydactyly
- Camptodactyly
- Polydactyly
- Syndactyly
- Enchondromatosis (Ollier)
- Fibrous dysplasia
- usual form (Jaffe-Lichtenstein)
- with skin pigmentation and precocious puberty (McCune-Albright)
- Gaucher’s
- Hypophosphatemic rickets
- Marfan’s
- Multiple hereditary exostoses
- Neurofibromatosis
- Osteogenesis imperfecta
- Osteopetrosis
- (with osteopetrosis, you get pyknodysostosis for free)
- Osteopoikilosis
I dropped this list into some anagram maker software, and it burped out over 300 possible mnemonics. Among the more intriguing were: COMMON HEAD FOG (very common after studying dysplasias), COMMON DEAF HOG (we all feel like this at times), HO! COG OF MADMEN (senior residents approaching boards time), and MACHO MEN OF GOD (most people never get this confident about dysplasias). So, I leave it to the reader to choose the mnemonic that sticks in your brain the best.
Having regurgitated this list of possibilities, it is time to check and see if the gods are smiling, and our patient has one of these entities. Unfortunately, to do this, you actually have to learn something about these entities, so gird your loins and do so. Listed below are a few germane facts about each of them to get you started.
Achondroplasia
First, a few words on dwarfism. Most types are very rare and quite a few are lethal. Of the nonlethal types, the only really common type of short-limbed dwarfism is achondroplasia. Therefore, once you have learned the important stuff about achondroplasia, the smart money suggests moving on to more practical topics.
Classic achondroplasia is a common autosomal dominant disorder, and is compatible with a long life span. The homozygous form, born of two parents with this common heterozygous form, is quite rare and quite lethal. However, most cases of achondroplasia are due to new mutations, rather than inheritance from a parent.
The very name of this syndrome suggests that the primary problem here is a generalized defect in enchondral bone formation. Once you know this, you can predict a lot of the findings seen in these people. Most of the appendicular skeleton is formed by and grows in size by enchondral bone formation. Therefore, we can accurately predict that the long bones (and therefore the patient) will be short. The characteristic shape of the skull and face in achondroplasia are also a logical extension of these principles. The calvarium is modelled on membranous bone, and its eventual size is merely a reflection of brain size. These people have brains of normal size, so their calvaria are likewise of normal size. However, the face and skull base come from enchondral bone, and end up relatively small, in comparison to the skull. The foramina of the skull base and spine and the spinal canal are often small, which may lead to prominent neurological problems and spinal stenosis.
Cleidocranial Dysostosis (aka cleidocranial dysplasia)
This is an autosomal dominant disorder whose very name tells us a lot about it. Dysostosis indicates an abnormality in the development of bone, and cleido- (clavicle) and cranial (head) tell us where major abnormalities occur. This disorder occurs in both membranous and enchondral bone, and has a striking propensity for affecting midline structures. If you painted a big, broad stripe down the midline with a paintbrush from skull to groin, you’d paint over a lot of structures involved with this syndrome. Prominent features include a large head with delayed suture closure, Wormian bones, hypertelorism, a small face, dental dysplasia, hypoplasia or aplasia of the clavicles, a narrow pelvis, and several varieties of spinal abnormalities. Just about every other bone in the body may be involved as well, including the ossicles of the ear. Despite the midline tendency, the appendicular skeleton is also frequently involved.
These patients have a normal life expectancy. Prominent complications of this syndrome include dental anomalies, hearing loss, scoliosis, and dislocations of the shoulder, radial head or hip.
Dactyly
Various abnormalities of the fingers may be seen, either alone or in association with other findings in a variety of syndromes. If you spot odd-looking fingers, characterize them and then look in Drs. Taybi and Lachman’s book for zillions of possible causes. How do you characterize them? English will do — short fingers, fused fingers, and too many fingers are common varieties. However, if you want to look these up in a gamuts book, you first have to translate this to Doctor Talk(tm). Ergo, a short glossary follows:
| English | Doctor Talk |
| Short fingers | Brachydactyly |
| Too many fingers | Polydactyly |
| Two or more fingers are fused together | Syndactyly |
| Contractures of fingers | Camptodactyly |
| Inclined fingers, usually fifth | Clinodactyly |
| Long, spider-like fingers | Arachnodactyly |
Enchondromatosis
Most enchondromas are solitary. However, some unfortunate patients may have a syndrome of multiple enchondromas, a.k.a. Ollier’s syndrome. This syndrome, unlike MHE (see below) is not hereditary. Although any bone may be involved, the smart money is usually on the tubular bones of the arm and leg. Along with the classic central expansile pattern seen with classic solitary enchondromas, one may also see linear or columnar lucencies in the metaphyses, representing columns of growing cartilage. The main significance of this disorder is that some lesions will undergo malignant degeneration (5 – 30 %), usually to chondrosarcoma. This likelihood is even higher (approaches 100 %) when multiple enchondromas are associated with soft tissue hemangiomas (Mafucci’s syndrome). These hemangiomas may contain phleboliths, making the diagnosis possible on plain radiographs.
Fibrous Dysplasia
This idiopathic disorder is due to excessive proliferation of the spindle cell fibrous tissues in bones. Although 2 cases of a congenital autosomal recessive form of fibrous dysplasia have been reported, every other case has been sporadic, without any known hereditary component. Although this process may occur rarely in the cortical bone, the vast majority of cases originate in the medullary space. Therefore, most cases present as bony enlargement with the process seeming to arise from an expanded medullary space.
The main clinical significance of this entity depends upon exactly which bones are affected. These bones will exhibit deformity, enlargement, and pain. Occasionally, pathological fractures will develop, and malignant transformation to osteosarcoma is seen rarely (< 0.5 %).
Two forms of fibrous dysplasia are seen in general radiologic practice: the conventional form (Jaffe-Lichtenstein syndrome) which may be monostotic or polyostotic, and a polyostotic form associated with precocious puberty and café au lait spots (McCune-Albright syndrome). In the later form, the bony involvement is often unilateral, but not always.
Gaucher’s Syndrome
This familial disorder has no gender predilection, and often occurs in Ashkenazi Jews. The usual form of the disorder is associated with a normal life span, although infantile and juvenile forms may result in mental retardation and an early demise. Gaucher’s is also called sphingolipidosis, oddly enough, because sphingolipids tend to accumulate in the reticuloendothelial cells. Hold that thought: stuff accumulating in the RE cells and causing problems. For radiologists, it’s not so important what is accumulating as where it accumulates. As far as most radiologists are concerned, it could just as easily be chocolate chip cookie dough that is piling up in the RE cells. If you then consider where the RE cells hang out, you know where to look for abnormalities in the patient with Gaucher’s disease: the liver, the spleen, and the bone marrow. The liver and spleen are usually quite enlarged, as are the RE cells in the bone marrow (sometimes called Gaucher cells). The next concept to consider is that the marrow space in the bone is a closed space. As these Gaucher cells enlarge, the intramedullary pressure begins to rise, which eventually may lead to occlusion of the intramedullary veins and hence bone infarction. As these bone infarcts evolve, one will be able to see the typical findings on MRI and then other imaging methods. The osteonecrosis may also develop in a subchondral location such as the femoral head in about half of patients, leading to subchondral collapse and early arthrosis. These patients may also exhibit other osseous findings, including the so-called “Erlenmeyer flask” deformities of the femoral metaphyses. These widened metaphyses may be seen in 40 – 50 % of patients, and may be due to the marrow packing of the Gaucher’s cells. These patients may also be at increased risk for osteomyelitis.
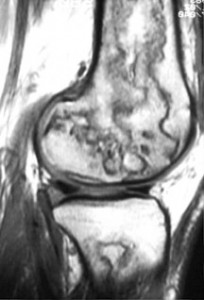
sagittal T1-weighted image of the knee — this image demonstrates multiple segmental areas of osteonecrosis in the distal femur in this patient with Gaucher’s syndrome
Hypophosphatemic Rickets
This has also been called X-linked hypophosphatemia, primary renal hypophosphatemic rickets or familial vitamin D-resistant rickets. As one of these names implies, it is due to a hereditary defect of the renal tubules, leading to decreased reabsorption of phosphate and therefore reduced serum phosphate levels. As the name also implies, this decreased reabsorption does not respond to usual amounts of vitamin D. This defect is passed on with an X-linked dominant mode of inheritance.
In general, this disorder exhibits rachitic epiphyseal and metaphyseal abnormalities predominantly in the lower limbs. This is best seen when comparing knee and wrist radiographs in the same patient. These patients also may demonstrate a generalized bone modeling error resulting in short, squat bones.
Marfan’s Syndrome
This is a familial disorder of connective tissue, primarily involving the eye, skeleton, and cardiovascular system. Although sporadic cases occur, most cases are due to autosomal dominant gene with a high degree of penetrance. I would like to be able to point to a single genetic defect of connective tissue that logically and inexorably leads to the findings in this syndrome. Alas, I can’t. Investigators are still uncertain of whether the primary defect lies in collagen, elastic fibers, or both. The skeletal overgrowth characteristic of this disorder is especially puzzling, and none of the current theories of defects in collagen synthesis do a very good job of explaining this overgrowth.
With this build-up, you would expect these patients to be characteristically tall and thin. You would be right. The limbs are disproportionately long with respect to the trunk, especially in the hands and feet, giving the appearance of “arachnodactyly”. These subjective impressions of the patient may be objectified somewhat by looking for the “thumb sign” (the thumb protrudes beyond a clenched fist), and measuring the segmental index (distance from pubic symphysis to floor / distance from top of head to floor) and the metacarpal index (length / width). Other common skeletal findings include scoliosis and hypermobile joints.
Common ocular abnormalities include bilateral ectopia lentis, myopia, and retinal detachments. Associated cardiac abnormalities lead to a shortened life expectancy for these patients. These abnormalities include cystic medial necrosis of the aorta or pulmonary arteries (leading to dissection or rupture), aortic and mitral insufficiency, a “floppy valve” syndrome, and septal defects.
Multiple Hereditary Exostoses
Osteochondromas usually have an absolutely pathognomonic appearance. The key word here is continuity. The cortex and medullary space of normal bone flows continuously into that of the osteochondroma (see figure below).
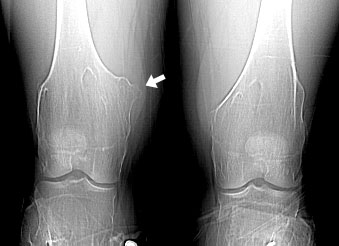
Multiple hereditary exostosis (MHE) syndrome is characterized by multiple osteochondromas throughout the skeleton, and this disorder seems to be hereditary (surprise!). Unfortunately, this syndrome is associated with an increased likelihood (up to 10 %, depending upon which study one quotes) that one or more of these osteochondromas will undergo malignant degeneration into something awful, usually a chondrosarcoma. Some patients without this syndrome will occasionally develop one or more osteochondromas. The likelihood of malignant degeneration is much lower (< 1 %) with sporadic osteochondromas such as this.
As it turns out, this fascinating syndrome has many points of similarity with some of the colonic polyposis syndromes. For example, the lesions may be pedunculated or sessile, they may be single or too numerous to count, and they may undergo malignant degeneration. The trick, then, in both syndromes, is determining which folks have the syndrome and which ones don’t. The answer in both cases is that one looks for evidence that the process is a systemic disorder and not just a focal, sporadic lesion. In the polyposis syndromes, one may look for other manifestations, such as associated osteomas (Gardner syndrome) or buccal pigmentation (Peutz-Jegher syndrome). With MHE syndrome, one looks for other dysplastic bones. A very common place to find this is in the femoral and humeral necks. Patients with MHE generally have short, thick necks in both anatomic sites. For this reason, after finding the first osteochondroma, one of the first things I want to see is a radiograph of the patient’s hips.
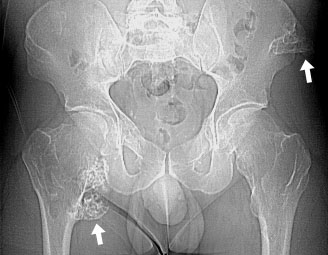
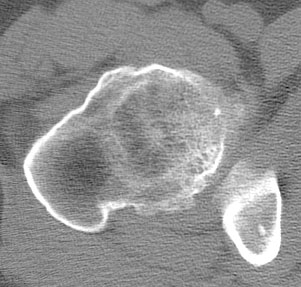
The radiograph and the CT demonstrate short, widened femoral necks. Also seen are several large osteochondromas (arrows).
You can explain a lot of things about osteochondromas if you consider them to be an ectopic epiphysis. This means that they grow right along with the normal epiphyses, and stop growing when the plates close. They look just like physes on radionuclide images in kids — hot until the plates close. These ectopic growth plates also sometimes will cause the bone to grow into strange shapes: too short, too long, or curved.
Once you have diagnosed this disorder, you must make sure that the patient knows the significance of their disorder and that they are now on a lifelong surveillance program. Any development of pain or growth after the plates have closed in an osteochondroma should be looked upon with suspicion for malignant degeneration. Followup imaging studies may include both radiographs and radionuclide images.
Neurofibromatosis
This disorder is seen in approximately one out of 3000 births. It is usually inherited as an autosomal dominant disorder, but there is a high rate of new mutations. There are jillions of reported manifestations, and virtually every part of the body is affected. Café au lait spots may be seen. There are two distinct forms of neurofibromatosis: the peripheral form (von Recklinghausen syndrome — seen in 90 % of patients) and the central form (acoustic neurofibromatosis).
The skeleton will be affected in about 80 % of patients. The most dramatic findings are the multiple neurofibromas seen throughout the body, especially in the peripheral, cranial, or spinal nerves and in the subcutaneous tissue. Fifty percent of patients with this disorder may develop kyphoscoliosis, usually in the high thoracic spine. This deformity may progress quite rapidly, and may lead to paraplegia. Other skeletal manifestations include posterior scalloping of vertebral bodies, hemihypertrophy, pseudarthrosis of the tibia, and enlargement of the spinal neural foramina.
Osteogenesis Imperfecta
This inherited, generalized disorder of connective tissue is characterized by abnormal maturation of collagen. It affects the skeleton, ligaments, skin, sclera, and teeth. The major clinical diagnostic triad is generalized osteoporosis with skeletal fragility, blue sclera, and odontogenesis imperfecta. Any two of these features suffice for the diagnosis.
Growth retardation occurs in most cases, and may be marked to the point of dwarfism in sever cases. This short stature is due to not only defects in collagen synthesis but also the cumulative fracture deformities secondary to the fragile bones.
The most common radiographic finding is that of generalized osteopenia. Multiple fractures resulting from insignificant trauma or normal muscle pull are also seen commonly, and may result in considerable deformity. Exuberant callus formation and pseudarthroses may also be seen. Persistent Wormian bones may be seen in the skull.
This entity should be considered when one is entertaining the diagnosis of the battered child syndrome, another cause of multiple fractures in multiple stages of healing. The workup of a potentially battered child is extremely serious, and involves significant legal and social investigations of the parents. It would be tragic to mistakenly invoke this massively invasive process on a family by missing the findings of osteogenesis imperfecta. The take-home message: always look carefully for other classic signs of osteogenesis imperfecta, such as generalized osteoporosis, Wormian bones, blue sclera and odontogenesis imperfecta.
Osteopetrosis
This is another very logical disorder. The prime defect may be a failure of osteoclastosis. Without properly working osteoclasts, the whole bone remodelling process will fare badly, leaving one with short, weak, and oddly shaped bones. With abnormal osteoclasts, one might predict the following abnormalities:
- The bones are very dense. In fact, they are so dense that nothing else really looks like this (except for pyknodysostosis, which is really, really rare).
- The bones fracture easily, often with a linear fracture plane.
- A proper medullary space is not created as the bones grow. Without a proper medullary space for the marrow, the patient will develop pancytopenia, leading to anemia (too few red cells), increased problems with infections (too few white cells), and bleeding problems (too few platelets). Because of these complications, few patients with this disorder survive childhood and the teens, unless they have the “tarda” form of the disease.
- The neural foramina may not grow as the patient and their nerves grow, leading to spinal or foraminal stenosis, especially at the skull base.
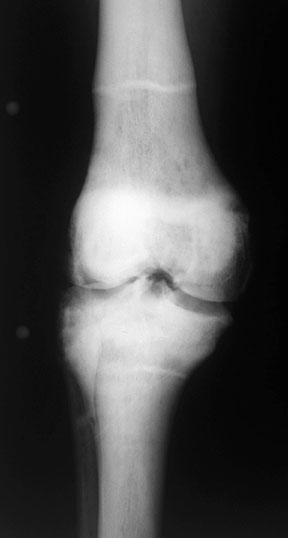

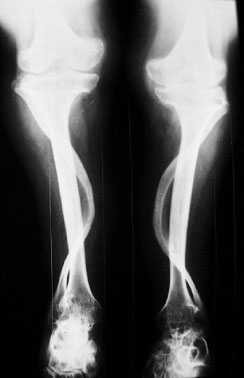
AP and lateral knee in a patient with osteopetrosis.
Osteopoikilosis
This disorder is considered to be very common. It is characterized by small round or oval foci of bone sclerosis located in the trabecular bone, particularly in the pelvis, metaphyses and epiphyses of long bones, tarsals, and carpals. The shoulders, hips and sacrum are especially good places to look for these findings. These little deposits of bone are essentially multifocal bone islands. Although some disagreeable things have been associated with osteopoikilosis (subcutaneous nodules, osteosarcoma, spinal stenosis, osteosclerosis, etc. ) these associated findings are probably pretty darned rare. The main clinical significance is that these may be mistaken for sclerotic metastases. Most of the time, their classic distribution and appearance will distinguish them readily from evil entities like mets.
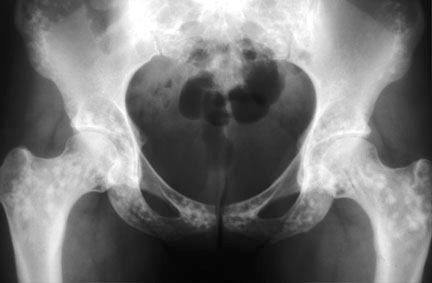
osteopoikilosis of the pelvis and proximal femurs
4. Could this dysplasia be in Taybi and Lachman’s Book?
Chances are extremely good that the syndrome is in Drs. Taybi and Lachman’s wonderful book, Radiology of Syndromes, Metabolic Disorders, and Skeletal Dysplasias. Besides providing a comprehensive list of syndromes, this book is also extremely well laid out. Each of the nearly 1000 entities in this book is listed the same way: its name, a list of synonyms, the mode of inheritance, frequency of occurrence, the clinical findings, the radiographic findings, a differential diagnosis (if any) and a list of pertinent references, including the original description(s) of the syndrome. This information is listed very efficiently (you have to, to get 1000 entities into only 800 pages). Thank goodness. When events have finally forced you to deal with the hand-foot-uterus syndrome once and for all, you pretty much just want to read the Cliff Notes version — not all 34 Cantos of Dante’s Inferno. Furthermore, this book is cross-referenced by means of lengthy gamut tables in the back of the book. If you can spot several key findings in the patient, you may be able to triangulate down onto a fairly short differential diagnosis or even one best diagnosis. If this process leads you to a certain diagnosis, life is good.
If you don’t have access to a copy of Taybi and Lachman’s book, you can go online to a wonderful website known as the Online Mendelian Inheiritance in Man (OMIM) Database. This site is essentially a constantly-updated textbook on congenital disorders of all types. As of March 29, 2012, they listed 21,153 entries, which should provide a long enough differential diagnosis for almost anyone.
If you can’t find it in Taybi and Lachman or in OMIM, you have done your best, and it is probably now time to punt.
5. Could it be time to call a real expert?
If you have to ask the question, the answer is yes. Remember that there is no shame in this — when it comes to dysplasias, virtually everyone feels like a first year resident. Refer early and often.
The next question is, how do you do this? What if you don’t know anyone that is a dysplasia expert. What if you don’t have one in your entire town, state or country?
The first rule here, is:
Don’t. Call. Me.
The reason for this is simple — I’m not a real dysplasia expert, I just play one on TV.
Seriously, while I am expert in many areas of musculoskeletal radiology, dysplasias are not one of my areas of expertise. This online chapter is my own meatball approach to dealing with this difficult topic, and was written to help my radiology residents to get through the night. As a diagnostic radiologist, I don’t actually see patients in clinic and I don’t do any sort of treatment.
How do you find a Real Dysplasia Expert®? Here’s what I do: I run a PubMed search on the disorder of interest and find out who’s been writing all of the papers on that disorder. These are the real experts. It’s entirely possible that the greater the number of papers written by someone, the bigger the expert they are. The Index Medicus citations from PubMed also list the institution where these experts work. You can then use the web or an actual phone book to find contact information for them. Hopefully, there will be one in a town, state or country near yours.
References:
- Potter B. The tale of the flopsy bunnies. New York: Frederick Warne, 1909:85.
- Taybi H, Lachman RS. Radiology of syndromes, metabolic disorders, and skeletal dysplasias. (4th ed.) Chicago: Year Book, 1996.
- Richardson ML, Helms CA, Vogler JB 3d, Genant HK. Skeletal changes in neuromuscular disorders mimicking juvenile rheumatoid arthritis and hemophilia. AJR 1984;143:893-897.
- Kozlowski K, Beighton P. Gamut Index of Skeletal Dysplasias : An Aid to Radiodiagnosis. Berlin: Springer-Verlag, 1984:182-189.
- Beighton P, et al. International Nomenclature of Constitutional Diseases of Bone. Revision, May, 1983. Ann Radiol (Paris) 1983 Sep-Oct;26(6):457-62.